Copy Number Inference From Exome Reads
CoNIFER uses exome sequencing data to find copy number variants (CNVs) and genotype the copy-number of duplicated genes. As exome capture reactions are subject to strong and systematic capture biases between sample batches, we implemented singular value decomposition (SVD) to eliminate these biases in exome data. CoNIFER offers the ability to mix exome sequence from multiple experimental runs by eliminating batch biases. Together with a short read aligner such as mrsFAST which can align reads to multiple locations, CoNIFER can robustly detect rare CNVs and estimate the copy number of duplicated genes up to ~8 copies with current exome capture kits. To get started right away, read the quick start guide.
News:
| 07.11.2011 | Critical CoNIFER v0.2.2 Update All CoNIFER users should update to this version of CoNIFER, as it fixes a bug in the RPKM algorithm where all bam files were considered to have a fixed number (10,000,000) of reads. This upgrade may change the output from outputs generated using version v0.2.1, but should be compatible with earlier versions. CoNIFER can be downloaded from HERE. |
| 07.11.2011 | CoNIFER v0.2.1 Update This is primarily an incremental update and fixes a variety of issues, especially pertaining to contig and probe naming. In addition, a new option to visualze the scree plot has been added (use --plot_scree filename.png). It can be downloaded from HERE. |
| 05.17.2011 | New CoNIFER Version Released (v0.2) It can be downloaded from HERE. This version has completely overhauled the workflow and pipeline for CoNIFER. All of the analysis is now done in a single step, and all the data is conveniently stored in a single HDF5 container file. Additionally, new plot routines have been added for better visualization of the results. This version of CoNIFER is the first beta release and is subject to the GPL license. See the updated quickstart and tutorial pages for more information. |
| 05.15.2011 | CoNIFER update to be released this week Based on feedback from initial alpha and beta testers, a new version of CoNIFER will be released this week. The new version will be significantly faster, streamline processing, and improve data export and graphing options. |
| 05.14.2011 | CoNIFER manuscript now available at Genome Research Advanced Online Publication!
Krumm et al. 2012. Copy number variation detection and genotyping from exome sequence data. Genome Research, doi:10.1101/gr.138115.112 |
How does CoNIFER work?
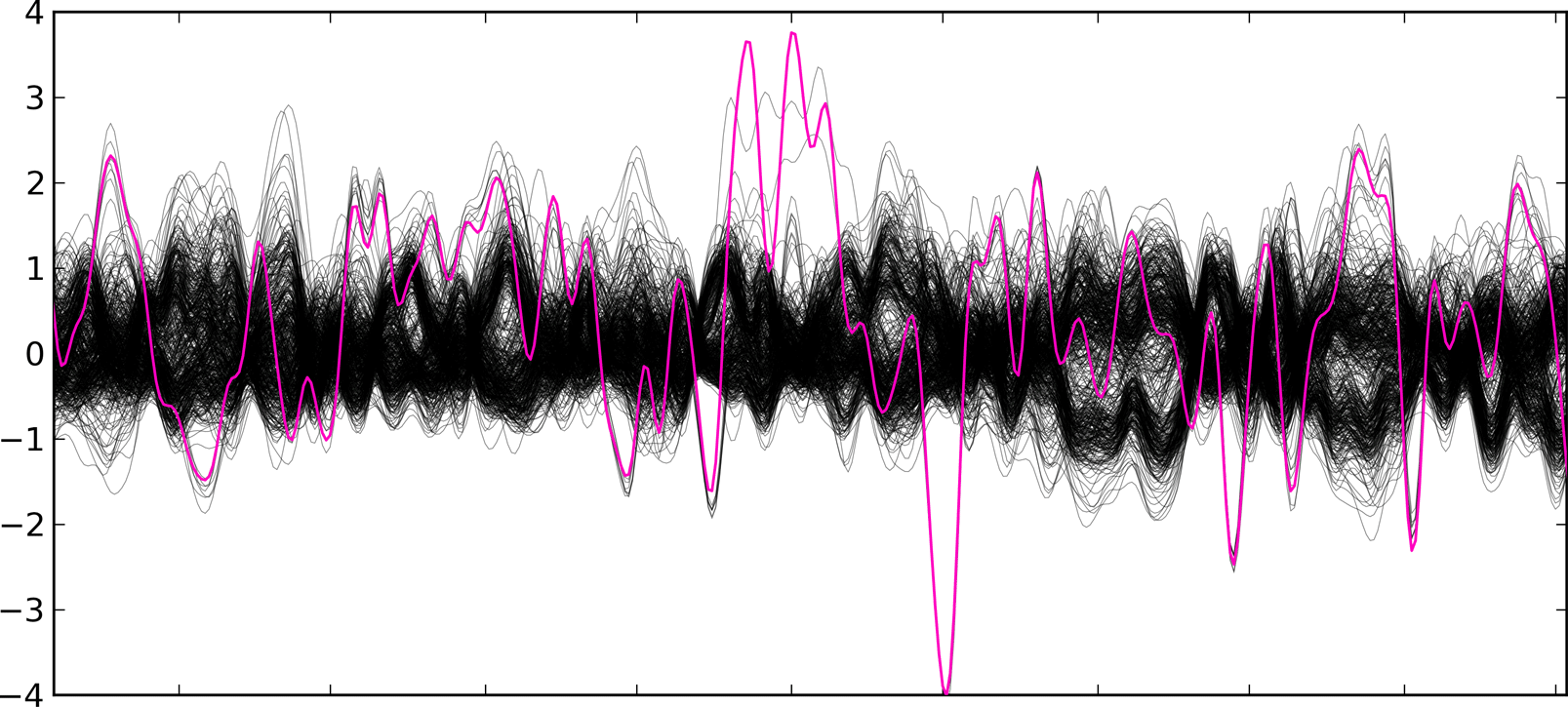
Briefly, CoNIFER calculates the number of sequencing reads that align to exons and then calculates a normalized RPKM (Mortazavi et al.) value. Each RPKM value is normalized by median and standard deviation across the analyzed population.
The Z-RPKM values are inputted into the SVD transformation (try interactive SVD demo), which removes systematic bias. The final SVD-ZRPKM signal is then smoothed and the duplication/deletion breakpoints are found using a threshold algorithm.
Can I use it?
Yes! CoNIFER is currently implemented as a small collection of python scripts availabe for public use. Head over to downloads to get the latest version. CoNIFER requires Python version 2.6 or later, and requires at minimum the numpy library. Additionally, pysam is required to calculate RPKM values from bed files and matplotlib is required for graphing and plotting. For more details, see the quick start guide or the tutorial.
More Resources
CoNIFER Sourceforge page
Submit Bugs and Feature Requests
MrsFAST Short Read Aligner